Do Deep Learning Models Really Outperform Traditional Approaches in Molecular Docking?
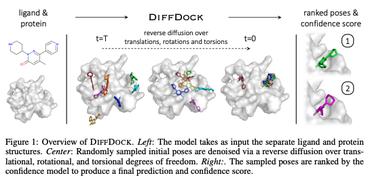
Molecular docking, given a ligand molecule and a ligand binding site (called ``pocket'') on a protein, predicting the binding mode of the protein-ligand complex, is a widely used technique in drug design. Many deep learning models have been developed for molecular docking, while most existing deep learning models perform docking on the whole protein, rather than on a given pocket as the traditional molecular docking approaches, which does not match common needs. What's more, they claim to perform better than traditional molecular docking, but the approach of comparison is not fair, since traditional methods are not designed for docking on the whole protein without a given pocket. In this paper, we design a series of experiments to examine the actual performance of these deep learning models and traditional methods. For a fair comparison, we decompose the docking on the whole protein into two steps, pocket searching and docking on a given pocket, and build pipelines to evaluate traditional methods and deep learning methods respectively. We find that deep learning models are actually good at pocket searching, but traditional methods are better than deep learning models at docking on given pockets. Overall, our work explicitly reveals some potential problems in current deep learning models for molecular docking and provides several suggestions for future works.
PDF Abstract